
Mixed Connective Tissue Disease Presenting as Longitudinal Extensive Transverse Myelitis and Vasculitic Neuropathy
*Corresponding author: Anuj Tikoo, Junior Resident, Department of General Medicine, Government Medical College, Nagpur, Maharashtra, India. anujtikoo111@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Tikoo A, Deshpande A. Mixed connective tissue disease presenting as longitudinal extensive transverse myelitis and vasculitic neuropathy. Vidarbha J Intern Med 2022;32:83-5.
Abstract
Mixed connective tissue disease (MCTD) is described as an entity with mixed features of systemic lupus erythematosus, systemic sclerosis, polymyositis/ dermatomyositis, and rheumatoid arthritis together with the presence of high-titre anti-U1 small nuclear and anti-ribonucleoprotein (anti-RNP) antibodies. Here, we present a case of an 18-year-old female patient who presented with quadriparesis, sensory loss in all four limbs, and trophic ulcers. Laboratory investigations were strongly positive for ANA, KU, SM/RNP, SM, SSA, and RIBOSOME P protein. MRI brain showed diffuse T2 hyperintensity in the spinal cord extending from cervicomedullary junction to conus with a subtle expansion of cord. A diagnosis of longitudinal extensive transverse myelitis and vasculitic neuropathy in the case of MCTD was made.
Keywords
Mixed connective tissue disorder
Longitudinal extensive transverse myelitis
Peripheral neuropathy

CASE REPORT
An 18-year-old female patient presented to the Emergency Medical Services of GMCH, Nagpur, with complaints of fever, headache, and a rash all over the body for the past 6 months; loss of sensations and progressive weakness of all four limbs over a period of 1 month. This was associated with loss of weight, painful movement at joints, ulcers over the right foot and right elbow joint, hair loss, difficulty in swallowing, change in voice and generalised body ache. She had a history of amenorrhoea for the past 1 year. There was no history of seizures. There was no history of hypertension, diabetes mellitus, and tuberculosis.
On admission, the patient was irritable, emaciated, body mass index – 17.9 kg/m2, afebrile, pulse rate – 86/min, respiratory rate – 18/min, blood pressure – 100/70 mmHg, and mild pallor was present. There were no icterus, cyanosis, clubbing, or lymphadenopathy. Alopecia was present. She had pain on passive movements of the neck from side to side. Rash was seen in both upper and lower limbs and ulcers were present over the right foot and medial aspect of the right elbow. No spinal deformity or tenderness was present on palpation [Figures 1-3].
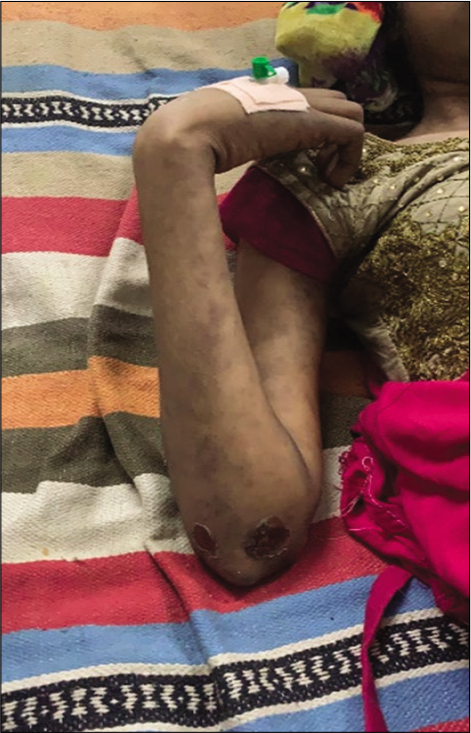
- Ulcers over the right elbow.
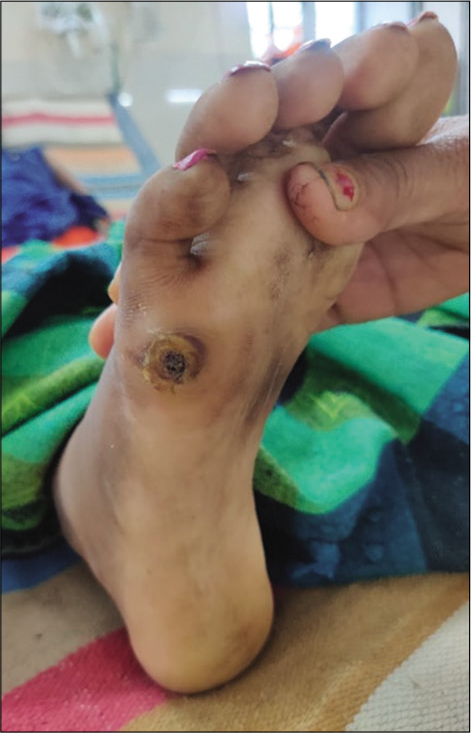
- Ulcers over dorsum of foot.
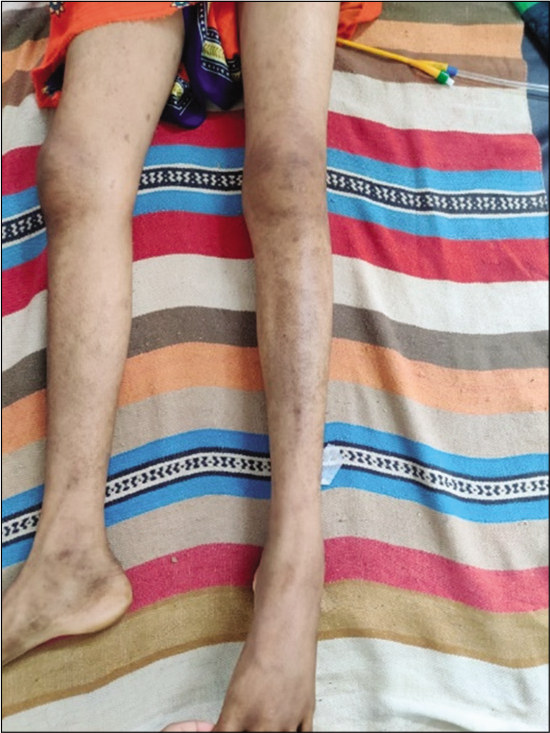
- Rash over both lower limbs.
Her neurological examination revealed generalised wasting, hypotonia of all four limbs, Grade 0 power in all four limbs, hypoesthesia below the level of C5 dermatome with exaggerated deep tendon reflexes and extensor plantars. However, there were no neck stiffness and no involvement of any cranial nerve. Abdominal system examination revealed ascites. Cardiovascular and respiratory system examinations were within normal limits.
Laboratory investigations revealed haemoglobin of 8.6 g %, TLC 18,100/ul, platelets 264,000/ul, blood urea 38 mg%, serum creatinine 0.5 mg%, sodium 135 mmol/l, potassium 3.7 mmol/l, total proteins 5.4 gm%, serum albumin 2.7 g %, total bilirubin 0.3 mg%, ALP 154 IU/L, ALT 42 IU/L and AST 80 IU/L.
Peripheral smear revealed a microcytic picture with moderate hypochromia. Her HIV, HBsAg, and HCV were negative. ANCA, RA factor, dengue, widal, and HRP2 were negative. hS-CRP was 4.5 mg%. CSF showed proteins of 208 mg/dl, low sugar values, 10 leucocytes (neutrophils 70% and lymphocytes 30%), ADA – 4.4 IU/L (normal <10 IU/L) and CSF CBNAAT negative. ANA, KU, SM/ ribonucleoprotein (RNP), SM, SSA, and ribosome P protein were strongly positive.
ECG 2D-ECHO and fundus were within normal limits. Nerve conduction studies revealed severe grade axonal motor and sensory polyneuropathy involving all four limbs.
MRI brain (Plain+ Contrast) with whole spine screening showed multiple small acute infarcts in bilateral frontoparietal cortical, subcortical at grey and white matter junction, heterogeneous enhancement in the right temporoparietal region, and diffuse T2 hyperintensity in cervicodorsal cord extending from cervicomedullary junction to conus with a subtle expansion of cord [Figures 4 and 5].
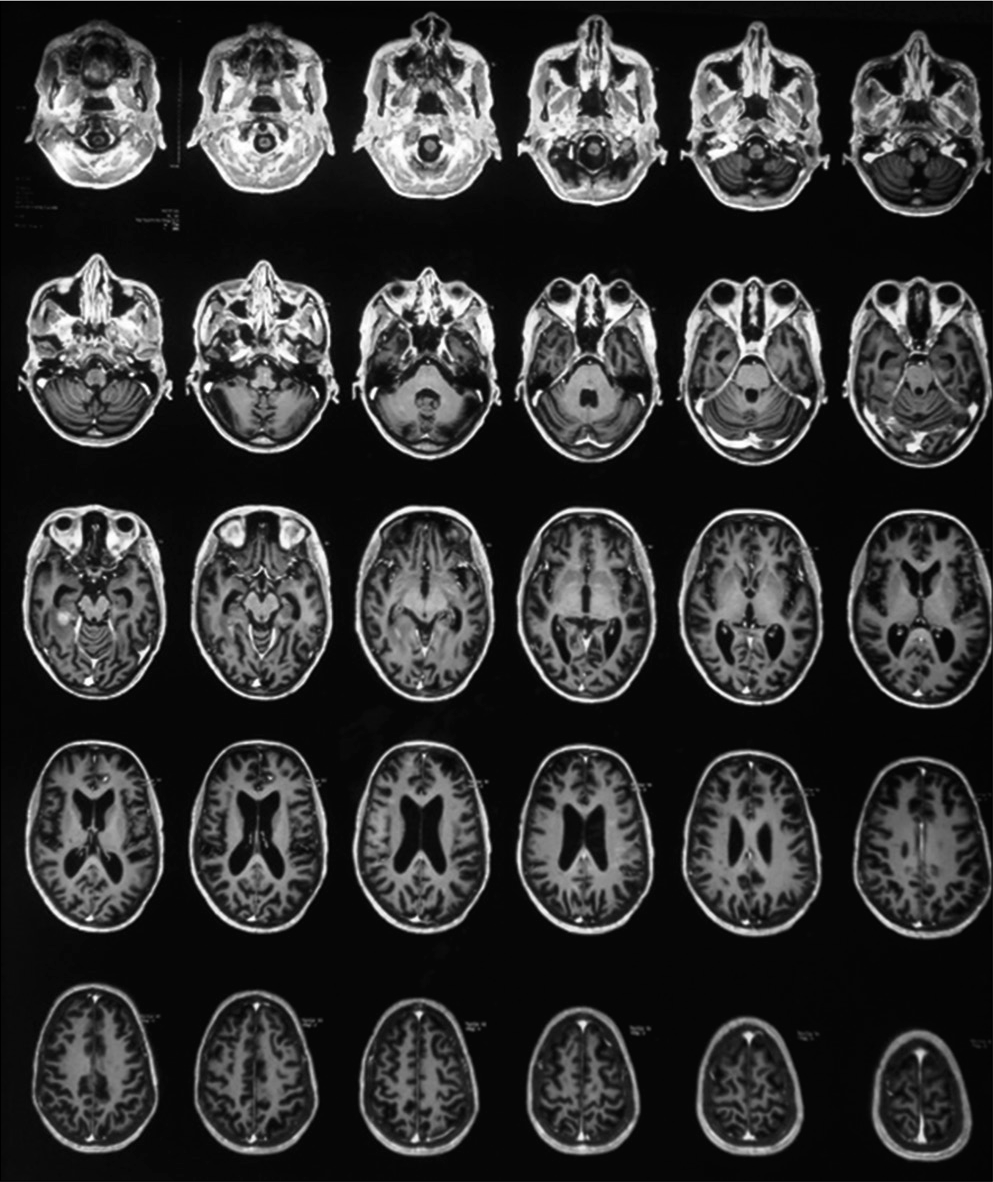
- Bilateral frontoparietal acute infarcts and hyperintensity over the right temporoparietal region.
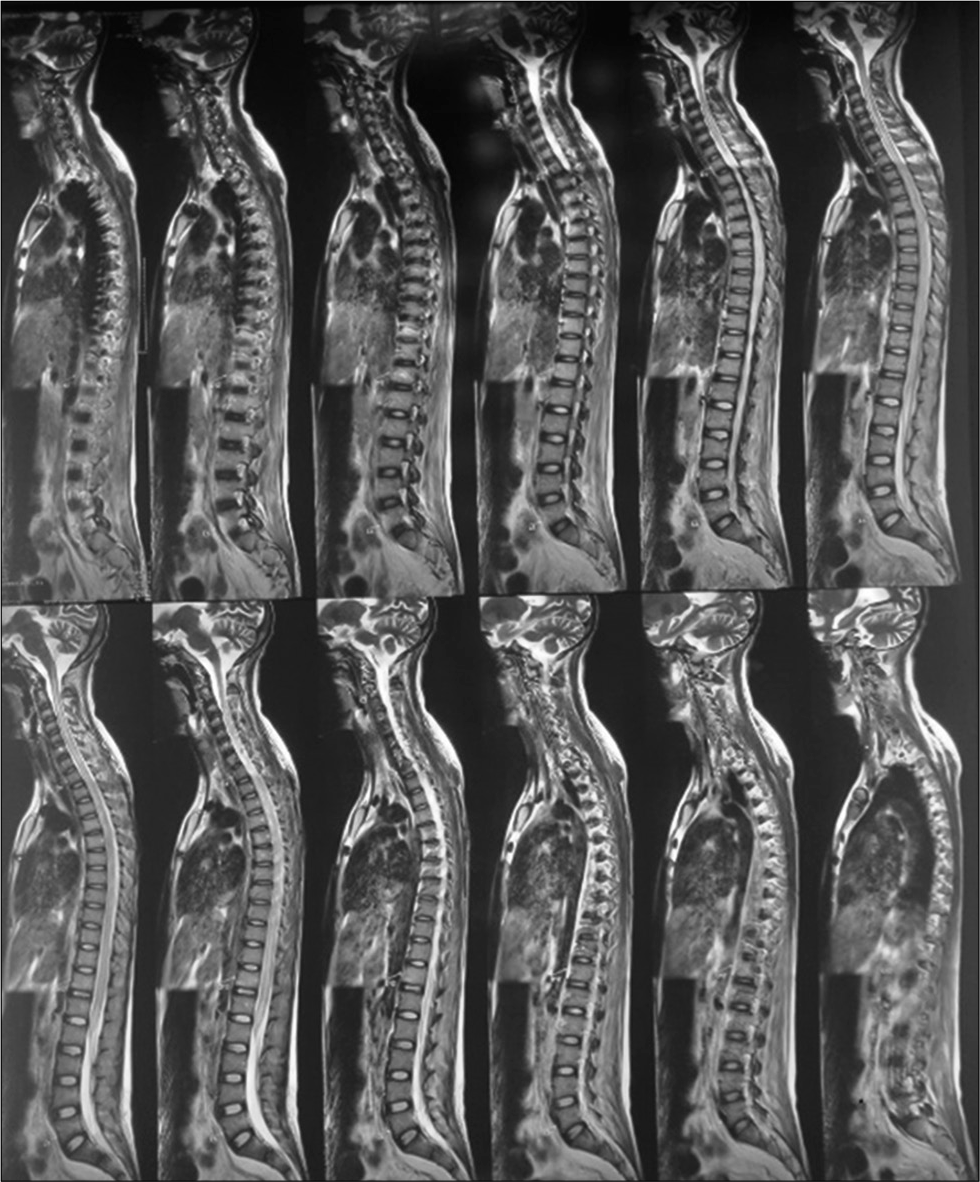
- T2 hyperintensity over cervicodorsal cord.
From the above history, clinical findings, and investigations, a diagnosis of mixed connective tissue disease (MCTD) with longitudinal extensive transverse myelitis and vasculitic neuropathy was made.
DISCUSSION
The term MCTD has been applied to a particular subset of patients with overlapping clinical features of lupus, scleroderma, myositis, and rheumatoid arthritis. The disease has been reported in children and is more prevalent in women in the third decade of life. MCTD has also been associated with HLA Class II alleles. HLA-DR4, DR1, and less frequent DR2 are the main HLA-associated alleles.[1,2] The most common clinical manifestations of the mixed connective disease are Raynaud’s phenomenon, arthralgia, swollen joints, oesophageal dysfunction, muscle weakness, and sausage-like finger appearance together with the presence of anti-RNP antibodies.[3,4] They appear in 90% of patients and usually develop insidiously.[5] Asthenia and myalgia are also frequent.
In all cases, therapy should be individualised for each patient to address the specific organs involved. Life-threatening manifestations require aggressive treatment. Treatment recommendations are corticosteroids (1 mg/kg daily). There should be a reduction of 5–10 mg every other week until a maintenance therapy using 5–10 mg prednisolone daily is achieved. If patients have severe neuropathy, high-dose corticosteroids (methyl prednisolone – 1000 mg daily) can be initiated for 3–5 days followed by prednisolone 1 mg/kg daily. Other modalities include IvIg at a dose of 2 g/kg body weight, rituximab, and cyclophosphamide (0.6–0.75 kg/sqm).
MCTD is a well-defined entity with a wide spectrum of clinical manifestations. Long-term studies reveal that some patients may have a mild self-limited disease, whereas others may develop severe major organ involvement. The worst prognosis and high mortality are associated with the presence of pulmonary arterial hypertension (PAH). MCTD can be a serious disease with the development of PAH, glomerulonephritis, vasculitis, gastrointestinal bleeding, and severe central nervous involvement, which does not always have a good prognosis.
Declaration of patient consent
Consent of Legally authorised Representative has been taken.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Analysis of anti-U1 RNA antibodies in patients with connective tissue disease. Association with HLA and clinical manifestations of disease. Arthritis Rheum. 1995;38:1837-44.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular genetic analysis of HLA-DR and HLA-DQ genes among anti-U1-70-kd autoantibody positive connective tissue disease patients. Arthritis Rheum. 1992;35:83-94.
- [CrossRef] [PubMed] [Google Scholar]
- Mixed connective tissue disease-an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA) Am J Med. 1972;52:148-59.
- [CrossRef] [Google Scholar]
- Mixed connective tissue disease. Arch Dermatol. 1976;112:1535-8.
- [CrossRef] [PubMed] [Google Scholar]
- The arthritis of mixed connective tissue disease. Ann Rheum Dis. 1987;37:397-403.
- [CrossRef] [PubMed] [Google Scholar]




