
Translate this page into:
Case of Parry-Romberg Syndrome: A Rare Case with New Presentation
*Corresponding author: Ruchi Sanjay Agrawal, Junior Resident, Department of Medicine, Government Medical College, Nagpur, Maharashtra, India. ruchiagrawal2737@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Agarwal RS, Kaur A, Khandait V. Case of Parry-Romberg syndrome: A rare case with new presentation. Vidarbha J Intern Med 2022;32:70-2.
Abstract
Parry-Romberg syndrome (PRS) is a rare clinical acquired slowly progressive disorder characterised by atrophy of one half of the face. The aetiology remains largely unknown. PRS has been associated with various neurological, ophthalmological, maxillofaciodental, and dermatological conditions. Here, we review a case of PRS and differentiate it from linear scleroderma en coup de sabre.
Keywords
Parry Romberg Syndrome
Linear Scleroderma

INTRODUCTION
Parry-Romberg syndrome (PRS), also known as progressive haemifacial atrophy, is a rare, acquired disorder characterised by progressive atrophy of the skin, subcutaneous tissue, and muscles of one side of the face and can also extend to cartilage and the underlying bony structures.[1,2] Alopecia and pigmentation of involved skin often appear.[3] PRS progresses over 2–20 years and then becomes stable.[4] Alternatively, the condition may burn out itself in a very early stage and results in minimal deformity.[5] Although the haemiatrophy is restricted to one half of the face, in rare cases, it can involve the same side arm, limb, and trunk. Furthermore, bilateral progressive atrophies have been reported.[4] There is a close relationship between PRS and linear scleroderma en coup de sabre (LSCS).[6]
CASE REPORT
A 12-year-old girl came to OPD with complaints of progressive flattening of forehead on the right side and sparse hair over eyebrows on the right side for 3 years as compared to the left leading to the asymmetry of the face. Her symptoms stabilised for the past 6 months. The patient also complained of patchy loss of scalp hair on the same side for 3 years for which she was undergoing treatment since then but with no improvement. Examination revealed obvious deformity of the right side of the face with atrophy of the skin and subcutaneous tissue over the forehead, thinning of the right eyebrow, and scarring alopecia on the affected side [Figures 1-3]. There was no induration, tightening, or tethering of the underlying skin. Ocular, ENT and neurological examinations were normal.
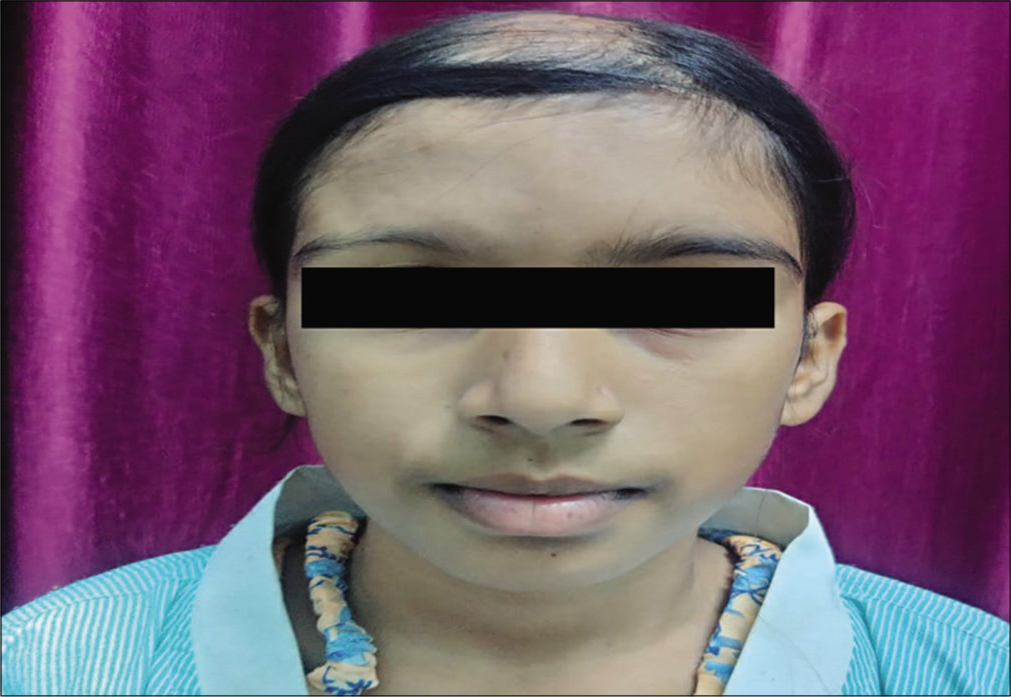
- Photograph showing atrophy of the right forehead and sparse eyebrow on the right side.
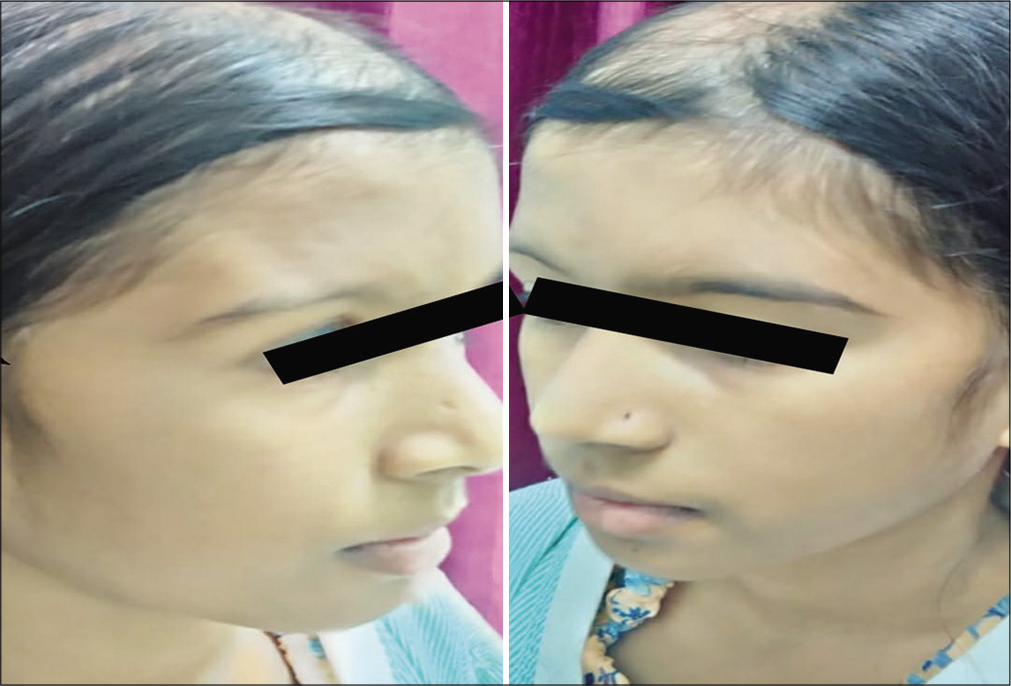
- Photograph showing atrophy of forehead and sparse eyebrow on the right side and right-sided alopecia compared to normal left side.
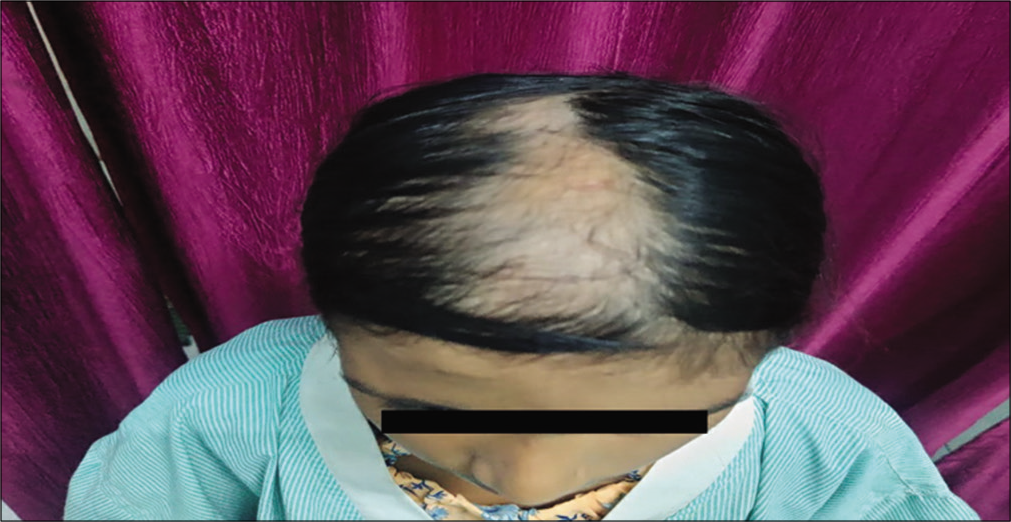
- Photograph showing alopecia limited to the right side with sparing of the left side.
Investigation
Routine blood investigations were normal. ANA by IFA was negative. CT head plain was normal and 3D CT of the skull revealed no involvement of the underlying bone. Biopsy was taken from midline scalp which showed atrophy of the epidermis, with marked collagenisation of the dermis without cellular infiltration consistent with morphological findings of PRS.
DISCUSSION
The incidence is of PRS which has been reported one in 700,000 in some studies.[7] The aetiology of PRS remains largely unknown and seems to be heterogeneous. Trauma, infection by slow viruses or bacteria, cranial vascular malformation, immune-mediated process, disturbances of fat metabolism, and sympathetic dysfunction have been proposed. The pathogenesis remains unknown.[8] The typical symptom is thinning and shrinkage of various tissue of the face which usually begins in the middle portion of the face such as the cheek or between the noses as the disease progresses, the upper face may be involved.[1-3] Other systemic manifestations include neurological and ophthalmological.[8]
The closest differential of PRS is LSCS.[6,9] Differentiating PRS from linear scleroderma is very challenging due to similar age of onset, similar lesions that progress overtime, both disorders have comparable neurological and ophthalmological manifestations. Some authors believe that they are separate entities while others endorse that progressive haemifacial atrophy is a part of scleroderma, a spectrum ranging from linear scleroderma through PRS to systemic sclerosis.[9] Histology of both PRS and scleroderma is similar that is, there is atrophy of the epidermis, dermis, subcutaneous tissue, skin adnexa, vessels, and/or hair follicles, and skin fibrosis with collagen.[8,9] However, in LSCS, there is massive lymphocytic infiltration around the vessels and deep plexus of the skin. In PRS, there is fibrosis, collagenisation of the dermis without inflammatory infiltration.[8] At present, there is no standard treatment algorithm for PRS. In severe and rapidly progressive PRS, immunosuppressive drugs such as methotrexate, corticosteroids, and azathioprine have been tried.[10] The actual treatment is surgery for the correction of residual deformities and restoration of facial asymmetry. Once the disease stabilises, cosmetic therapies including dermal fat grafts, autologous fat grafts, muscle flap grafts, free silicone injections,[1] and bone augmentations are available.[11]
CONCLUSION
A rare case of Parry Romberg Syndrome that needs to be differentiated from Linear Scleroderma.
Declaration of patient consent
Consent of Legally authorised Representative has been taken.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Parry-Romberg syndrome and Rasmussen encepha-litis: Possible association; clinical and neuroimaging features. J Neuroimaging. 2009;20:1-6.
- [Google Scholar]
- Parry-Romberg syndrome: Facial atrophy and its relationship with other regions of the body. Ann Plast Surg. 2009;63:457-61.
- [CrossRef] [PubMed] [Google Scholar]
- Parry Romberg syndrome: A case report and discussion. J Oral Maxillofac Pathol. 2012;16:406-10.
- [CrossRef] [PubMed] [Google Scholar]
- Progressive hemifacial atrophy-case report. Med Oral Pathol Oral Cir Bucal. 2006;11:112-4.
- [Google Scholar]
- An unusual case of hemifacial atrophy. Oral Surg Oral Med Oral Pathol. 1992;73:564-9.
- [CrossRef] [Google Scholar]
- Linear scleroderma “en coup de sabre” of the cheek. J Oral Maxillofacio Surg. 2003;61:1091-4.
- [CrossRef] [Google Scholar]
- Parry-Romberg syndrome: A global survey of 205 patients using the internet. Neurology. 2003;61:674-6.
- [CrossRef] [PubMed] [Google Scholar]
- A review of Parry-Romberg syndrome. J Am Acad Dermatol. 2012;67:769-84.
- [CrossRef] [PubMed] [Google Scholar]
- Parry-Romberg syndrome associated with localized scleroderma. Case Rep Neurol. 2010;2:57-62.
- [CrossRef] [PubMed] [Google Scholar]
- Parry Romberg syndrome: 7 cases and literature review. AJNR Am J Neuroradiol. 2015;36:1355-61.
- [CrossRef] [PubMed] [Google Scholar]




