
Translate this page into:
Distinct Profiles of Pulmonary Embolism: Insights from a Case Series
*Corresponding author: Gyanshankar Mishra, Department of Respiratory Medicine, Indira Gandhi Government Medical College, Nagpur, Maharashtra, India. gpmishra81@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Agarwal K, Mishra G, Munje R, Atram JS. Distinct Profiles of Pulmonary Embolism: Insights from a Case Series. Vidarbha J Intern Med. 2023;33:90-4. doi: 10.25259/VJIM_6_2023
Abstract
Pulmonary embolism (PE) is a serious and potentially life-threatening condition with significant diagnostic and therapeutic challenges. Here, we present a case series of three patients with PE, each with a distinct clinical profile and severity, to provide insights into the heterogeneity of this condition. The cases illustrate the importance of timely diagnosis and individualised management approaches for optimal patient outcomes. The study presents a comprehensive analysis of the clinical, electrocardiographic, and radiological findings and therapeutic interventions employed in managing the cases. Our report highlights the need for continued research and clinical awareness of this complex condition to improve understanding and management. This case series adds to the existing literature on the diverse clinical presentations of PE and emphasises the critical importance of tailored management strategies.
Keywords
Antiphospholipid antibody syndrome
Pulmonary thromboembolism
Lung Infarct
Computed tomography pulmonary angiogram
Hyperhomocysteinemia

INTRODUCTION
Antiphospholipid syndrome (APS) and hyperhomocysteinemia are complex disorders with diverse clinical and laboratory features, posing a diagnostic challenge to healthcare providers. APS is characterised by the presence of persistent antiphospholipid antibodies (aPLs) and is associated with thrombotic events and pregnancy morbidity, affecting the vessels of various tissues and organs. APS may occur in isolation (primary APS) or association with systemic autoimmune diseases such as systemic lupus erythematosus (secondary APS).[1,2] Hyperhomocysteinemia is another disorder that can cause a hypercoagulable state, leading to thrombotic events. The suggested pathophysiological mechanisms underlying the development of these disorders include disruptions in the kinetics of normal procoagulant and anticoagulant reactions on cell membranes and alterations in the expression and secretion of various molecules.[3]
The pulmonary system is a common target of these disorders, and pulmonary thromboembolic disease can often be the first manifestation of APS or hyperhomocysteinemia. In addition to pulmonary embolism (PE), other pulmonary manifestations of these conditions include pulmonary hypertension, adult respiratory distress syndrome, intra-alveolar haemorrhage, and primary thrombosis of lung vessels.[4] Early recognition and timely management of these pulmonary manifestations are crucial in improving patient outcomes.
This review aims to provide a comprehensive overview of APS and hyperhomocysteinemia, with a focus on their associated pulmonary manifestations and the current diagnostic and treatment options available. By enhancing our understanding of the clinical and laboratory features of these disorders, healthcare providers can improve clinical suspicion, timely diagnosis, and effective management of these conditions.[1-4]
CASE REPORTS
Case report 1
We present the case of a 28-year-old non-smoking male with no prior medical history who presented with cough and haemoptysis for four days before admission, progressively worsening shortness of breath, bilateral chest pain, and fever for four days. The patient was previously hospitalised seven months ago for chest pain radiating to the left arm, shortness of breath, and fever with chills and vomiting, where the echocardiogram showed mild tricuspid regurgitation, mild pulmonary hypertension, and normal coronary angiography. On current admission, the patient had an average build with a saturation of 67% on room air and tachypnoea with a respiratory rate of 32/min. The patient’s blood pressure was 120/90 mmHg. The ECG showed global T wave inversion in ii, iii, and Avf V1 To V6, and Two D echocardiography (2D-ECHO) showed right atrium (RA) and right ventricular (RV) dilatation.
Further, the evaluation showed that the patient had a positive APS with positive antiphospholipid immunoglobulin M (IgM), anticardiolipin IgM, and beta 2 glycoprotein IgM antibodies, as well as elevated homocysteine levels. Chest X-ray was suggestive of the right cavitary lesion [Figure 1]. The computed tomography pulmonary angiogram (CTPA) showed a complete non-contrast filling defect in the right main pulmonary artery consistent with pulmonary thromboembolism (PTE) and a partial filling defect in the segment branch of the medial basal segment of the left lower lobe [Figure 2]. The patient was treated with an injection of enoxaparin twice a day and showed improvement in breathlessness and cough. The D-dimer and N-terminal prob-type natriuretic peptide levels decreased when repeated after 5 and 10 days. The patient was discharged on the tablet dabigatran 110 mg twice a day and advised to follow up after two months and repeat antibody testing after 12 weeks.
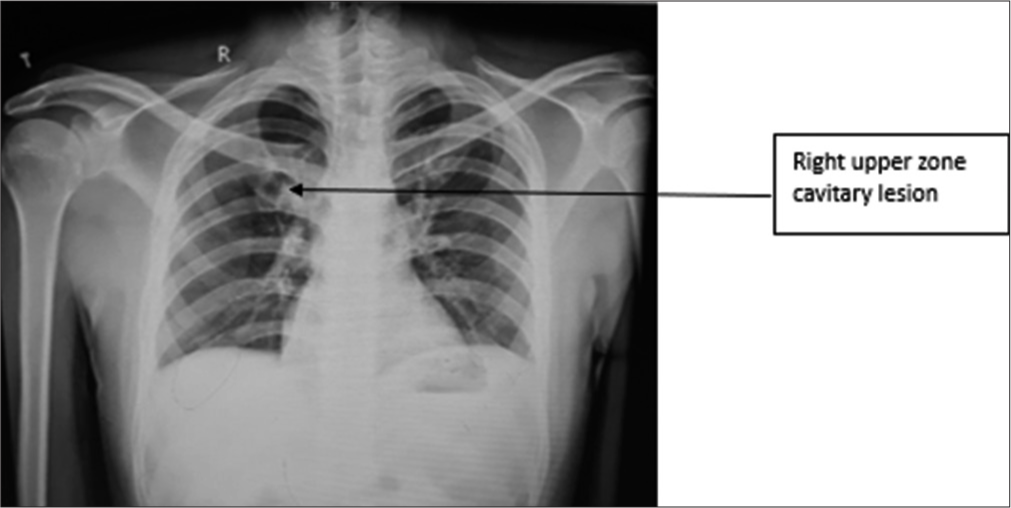
- Case 1 – Chest X-ray suggestive of the right upper zone cavitary lesion.
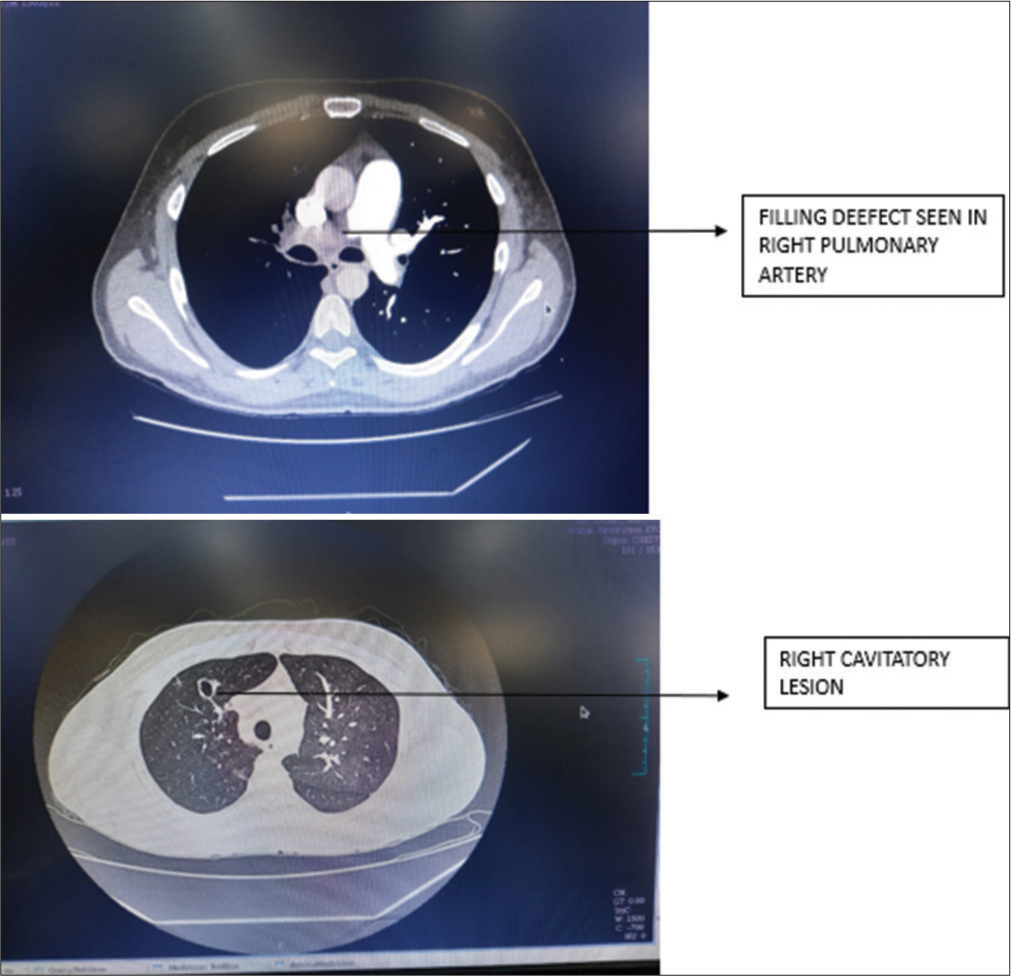
- Case 1 – Computed tomography pulmonary angiogram suggestive of filling defect in the right pulmonary artery and right cavitary lesion.
Case report 2
We present the second case report of a series of three cases, highlighting the clinical features, diagnostic workup, and pulmonary hypertension (PAH) in young adults.
A 32-year-old male driver presented with complaints of exertional breathlessness for six months, which had progressed from MMRC grade 0 to grade 4. He also had sudden onset increased breathlessness for one day before admission, cough for five days associated with whitish scanty expectoration, fever, and bilateral dull chest pain for three days. The patient denied any comorbidities or addictions. His pulse rate was 85/min, blood pressure was 120/80 mmHg, and saturation was 85% on room air.
On examination, the patient had tachycardia (135/min), and his chest X-ray showed left-sided oligemia with minimal left pleural effusion [Figure 3]. ECG showed right axis deviation. Routine blood investigations were suggestive of a raised total leukocyte count (18,000/mL), haemoglobin of 14 g/dL, and platelet count of 191,000/mL. Liver and kidney function tests were normal. D-dimer level was elevated (930 ng/mL). Based on these findings, PTE was suspected.
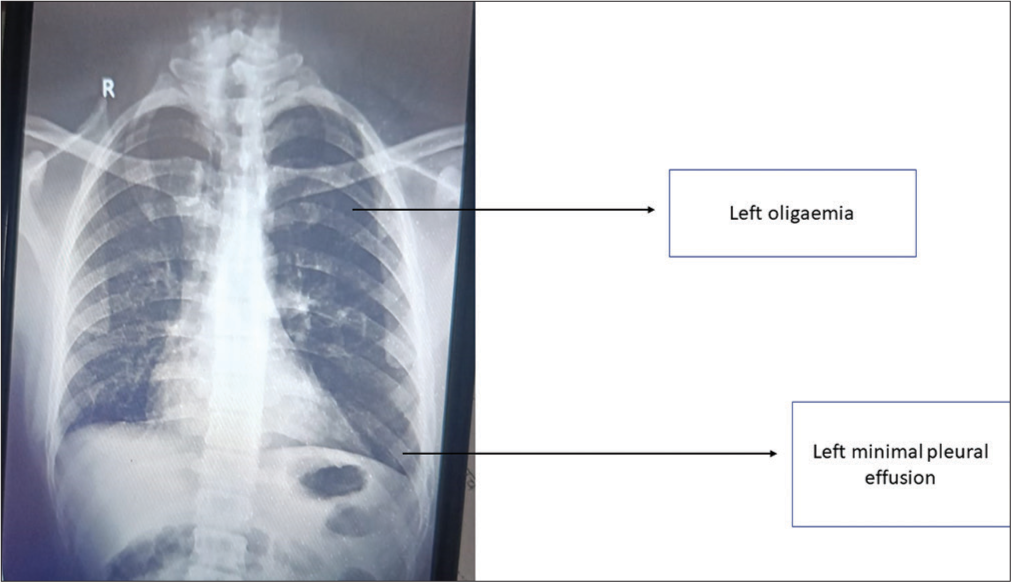
- Case 2 – Chest X-ray suggestive of the left oligaemia and left minimal pleural effusion.
The CTPA showed complete non-contrast non-opacification involving left pulmonary arteries extending into lobar and segmental branches [Figure 4] and reversal of aortopulmonary ratio (<1), suggesting changes in PAH. In addition, multiple cavitary and consolidatory changes with ground-glass opacities in the left lower lobe and minimal collection in the bilateral pleural space are shown in Figure 4.
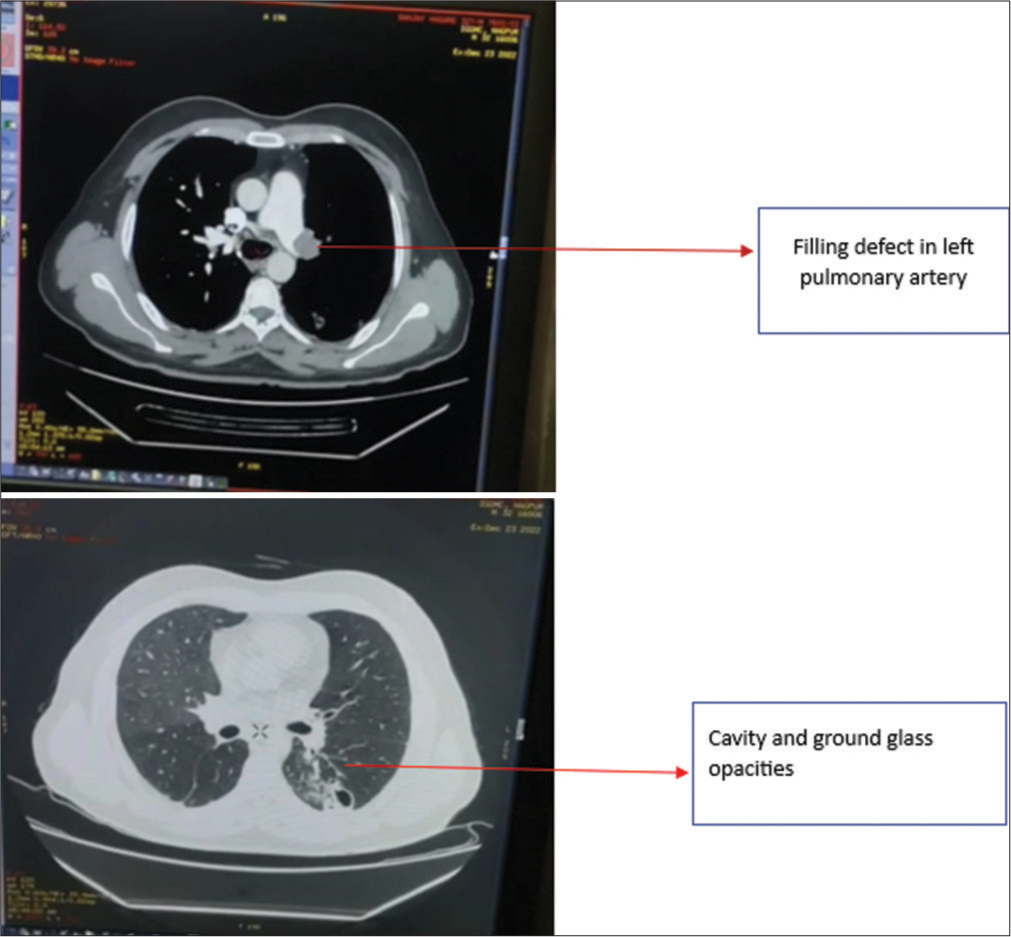
- Case 2 – Computed tomography pulmonary angiogram suggestive of filling defect in the left pulmonary artery and cavity and ground-glass opacities.
The patient’s bilateral upper and lower limb and carotid Doppler were normal. Sputum, human immunodeficiency virus (HIV), thyroid function test, and ultrasonography reports were also normal. Homocysteine level was elevated (25.1 mmol/L, normal <15.0). Protein S and protein C, lupus anticoagulant (LAC), antithrombin III, antiphospholipid IgM, cardiolipin IgG and IgM, and beta two glycoprotein IgM antibodies were all normal. 2D-ECHO was suggestive of severe PAH RA and RV dysfunction.
The patient was treated with antibiotics and enoxaparin. He was also given iron, vitamin, and folic acid supplements. The patient remained haemodynamically stable throughout hospitalisation, complained of exertional breathlessness, and did not experience any episodes of haemoptysis. He was admitted for 14 days and discharged on oral anticoagulants, antibiotics, and supplements, with a follow-up in 15 days.
Case report 3
A 26-year-old male with a history of occasional smoking and alcoholism presented with cough and expectoration for 45 days, which progressed to include yellowish expectoration with blood and breathlessness for 15 days. The patient also experienced left-sided chest pain for 15 days and intermittent, high-grade fever for three days before admission. On examination, the patient was haemodynamically stable with a pulse rate of 117/min and saturation of 97% on room air.
Chest X-ray revealed left-sided consolidation and left pleural effusion [Figure 5]. ECG showed tachycardia, and D-dimer was elevated at 2970 ng/mL. A CTPA showed a near-complete filling defect in the distal aspect of the left interlobar artery and a partial filling defect in the lateral basal and posterobasal segmental branch of the left pulmonary artery [Figure 6]. The CT scan also showed loculated left pleural effusion with underlying ground-glass opacities, consolidation, and cavitary changes [Figure 6].
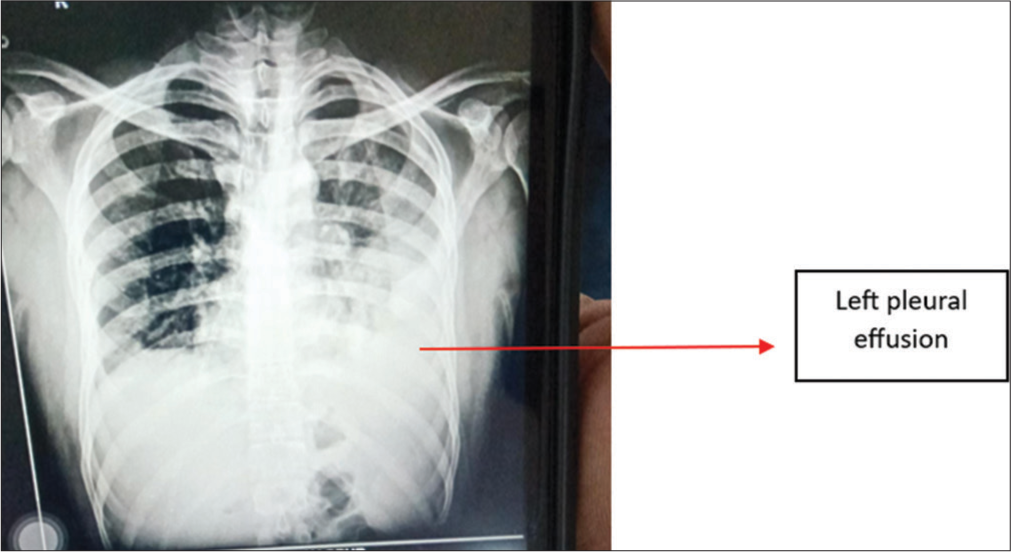
- Case 3 – Chest X-ray suggestive of the left pleural effusion.
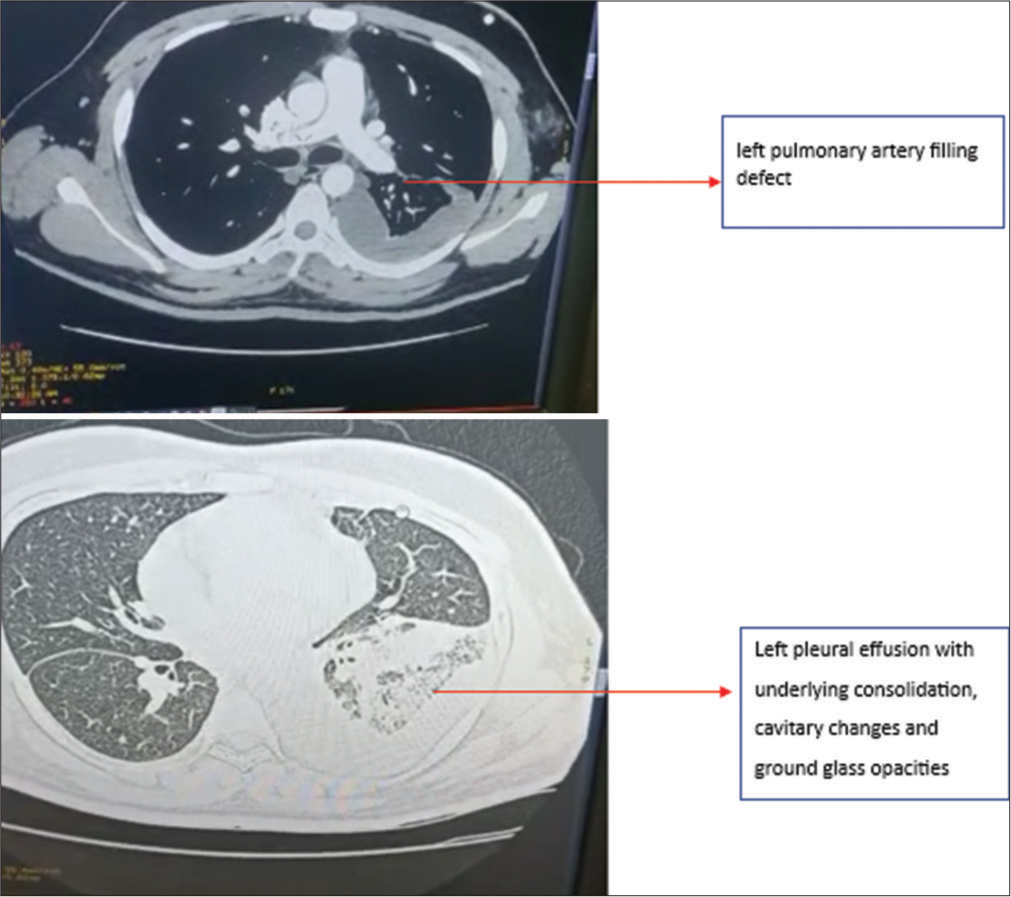
- Case 3 – Computed tomography pulmonary angiogram suggestive of the left pulmonary artery filling defect and left pleural effusion with underlying consolidation, cavitary changes and ground-glass opacities.
Further investigations, including HIV status, serum homocysteine, LAC, protein C and protein S, antithrombin III, anticardiolipin, beta two glycoproteins, antiphospholipid and antinuclear antibodies, were negative. Doppler studies of the bilateral upper limb, lower limb, and carotid were normal, and liver and kidney function tests were also normal. Pleural fluid was exudative with low adenosine deaminase levels. Ultrasonography of the abdomen showed mild hepatomegaly.
The patient was treated with low-molecular-weight heparin, pleural tapping, and intravenous antibiotics for 15 days and discharged on oral anticoagulants with follow-up instructions. Despite a full workup and detailed history, no cause of thromboembolism could be identified, and a diagnosis of idiopathic thromboembolism was inferred.
DISCUSSION
When a person experiences arterial or venous thrombosis, recurrent miscarriages, and laboratory testing for aPLs (anticardiolipin antibodies, or LAC, or both) are positive, the condition is known as the antiphospholipid syndrome.[4] APS tends to affect young to middle-aged women, with an estimated incidence of approximately five new cases/100,000 persons/year and a prevalence of around 40–50 cases/100,000 persons.[5] aPL may act in vivo by disrupting the kinetics of the normal procoagulant and anticoagulant reactions occurring on cell membranes.[3]
Hyperhomocysteinemia and thromboembolism have also been linked in studies, although this link may be complicated by lifestyle factors such as smoking status, body mass index, and physical activity.[6] The risk of recurrent venous thrombosis is almost three-fold higher in patients with hyperhomocysteinemia than in those with normal homocysteine levels.[6] Although a number of mechanisms, including increased tissue factor expression, attenuated anticoagulant processes, increased platelet reactivity, increased thrombin generation, increased factor V activity, reduced fibrinolytic potential and vascular injury, including endothelial dysfunction, elevated homocysteine levels are linked to thrombosis.[7]
Unprovoked thromboembolism is difficult to suspect and diagnose. Venous thromboembolism (VTE) is considered unprovoked when it occurs without known environmental risk factors in the absence of a prognostically important transient or a persistent provoking factor (such as cancer).[8] The prevalence of previously undiagnosed cancer in unprovoked VTE was 6.1% at the time of diagnosis.[8]
The management of acute thrombotic events in patients with APS, hyperhomocysteinemia, and unprovoked thromboembolism is the same as in the general population. Patients require anticoagulation treatment with heparin followed by warfarin. Long-term, possibly lifelong, oral anticoagulation is the optimum prophylactic treatment for recurrent thrombosis.[9]
These cases serve to underscore the rarity of PE presenting as the initial manifestation in young males while also emphasising the criticality of recognising the presence of both APS and hyperhomocysteinemia as potential causative factors. The identification of these hypercoagulable states is essential in determining the optimal therapeutic approach for patients with PTE, particularly those whose aetiology remains obscure despite conventional diagnostic evaluations.
CONCLUSION
Our case series underscores the significance of recognising antiphospholipid syndrome and hyperhomocysteinemia as potential aetiologies of VTE in young men. When traditional diagnostic methods prove inconclusive, exploring these hypercoagulable states should be considered. Identifying such conditions can significantly impact therapeutic outcomes, as anticoagulation therapy can effectively prevent recurrent thrombotic events. Nonetheless, the possibility of underlying connective tissue diseases must not be disregarded, as it can influence treatment approaches. In instances where a definitive diagnosis remains elusive despite extensive workup, idiopathic thromboembolism can be considered.
Ethical approval
The research/study complied with the Helsinki Declaration of 1964.
Declaration of patient consent
Patient’s consent is not required as patients identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- International Consensus Statement on an Update of the Classification Criteria for Definite Antiphospholipid Syndrome (APS) J Thromb Haemost. 2006;4:295-306.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical Spectrum of Males with Primary Antiphospholipid Syndrome and Systemic Lupus Erythematosus: A Comparative Study of 73 Patients. Lupus. 2004;13:11-6.
- [CrossRef] [PubMed] [Google Scholar]
- Antiphospholipid Syndrome: Pathogenic Mechanisms. Autoimmun Rev. 2003;2:86-93.
- [CrossRef] [PubMed] [Google Scholar]
- The Lung in the Antiphospholipid Syndrome. Ann Rheum Dis. 2002;61:195.
- [CrossRef] [PubMed] [Google Scholar]
- Epidemiology of the Antiphospholipid Antibody Syndrome. J Autoimmun. 2000;15:145-51.
- [CrossRef] [PubMed] [Google Scholar]
- Original Contribution Hyperhomocysteinemia and Risk of First Venous Thrombosis: The Influence of (Unmeasured) Confounding Factors. . 2023;187:1392-400.
- [CrossRef] [PubMed] [Google Scholar]
- Homocysteine and Thrombosis: From Basic Science to Clinical Evidence. Thromb Haemost. 2005;94:907-15.
- [CrossRef] [PubMed] [Google Scholar]
- Management of Idiopathic Venous Thromboembolism. Expert Rev Cardiovasc Ther. 2016;14:1371-84.
- [CrossRef] [PubMed] [Google Scholar]
- New Guidelines for the Diagnosis and Management of Pulmonary Embolism: Key Changes. World J Cardiol. 2020;12:161-6.
- [CrossRef] [PubMed] [Google Scholar]




