
Translate this page into:
An Interesting Case of Proteinuria: AL Amyloidosis
*Corresponding author: Dipti Chand, Department of Medicine, Government Medical College, Nagpur, Maharashtra, India. dachand.ngp@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Chand D, Gosavi R, Chakraborty A, Kungar T, Paliwal Y. An interesting case of proteinuria: AL amyloidosis. Vidarbha J Intern Med 2022;32:73-6.
Abstract
Light-chain (AL) amyloidosis is the most common form of systemic amyloidosis and is associated with an underlying plasma cell dyscrasia. The disease often is difficult to recognise because of its broad range of manifestations and what often are vague symptoms. The clinical syndromes at presentation include nephrotic-range proteinuria with or without renal dysfunction, hepatomegaly, congestive heart failure and autonomic or sensory neuropathy. Recent diagnostic and prognostic advances include the serum free light-chain assay, cardiac magnetic resonance imaging and serologic cardiac biomarkers. Treatment strategies that have evolved during the past decade are prolonging survival and preserving organ function in patients with this disease. This review outlines approaches to diagnosis, assessment of disease severity and treatment of AL amyloidosis. We describe a case of a 54--year-old male patient with monoclonal gammopathy with AL amyloidosis.
Keywords
Proteinuria
AL Amyloidosis
Cherry on the top

CASE REPORT
A 54-year-old male patient complained of chest pain, breathlessness and bilateral lower limbs swelling in January 2021, the breathlessness progressed gradually. He was evaluated in view of breathlessness, 2D echo was suggestive of mild pericardial effusion and concentric LVH, CT thorax was also done which showed ground-glass opacities in bilateral lower lobes. COVID RTPCR was negative. He was started on diuretics, swelling over his feet reduced to some extent but breathlessness persisted.
He again started to develop swelling over his legs; it was gradually progressive in nature, his breathlessness also increased in April 2021. Routine investigations were normal except for a low serum albumin which was 2.8 g/dl (3.2–4.8 g/dl). Renal parameters were also normal. Routine urine examination showed 3+ urine albumin and a raised urine/creatinine ratio of 2.84 (normal up to 0.15).
The patient was started on tab. enalapril in view of albuminuria. Kidney biopsy was done in view of nephrotic range proteinuria, which showed renal amyloidosis, glomerular and arterial and arteriolar deposition of amyloid, direct immunofluorescence showed dominance of lambda light chain staining over kappa light chains.
Bone marrow biopsy was done, which was normal; the patient was started on injection bortezomib and tab. thalidomide, he received one cycle of chemotherapy.
The patient presented to our OPD with the complaints of generalised swelling of the body and progressive breathlessness for 15 days. On examination, he was conscious, oriented; periorbital pigmentation was present, waxy look on the face and anasarca was present, blood pressure was 100/70, pulse was 90/min, regular, the patient also had purpuric spots over his body [Figure 1]. Systemic examination revealed basal crepitations and 5 cm hepatomegaly. Routine investigations were done, which revealed thrombocytopenia (49,000), serum creatinine was 1.6 mg/dl (0.5–1.2 mg/dl). Serum albumin was 2.3 g/dl and serum ionic calcium was 0.93 Meq/l (1.0–1.32). Troponin I was positive and NT-proBNP was more than 25,000 pg/ml. Remaining routine investigations were normal.
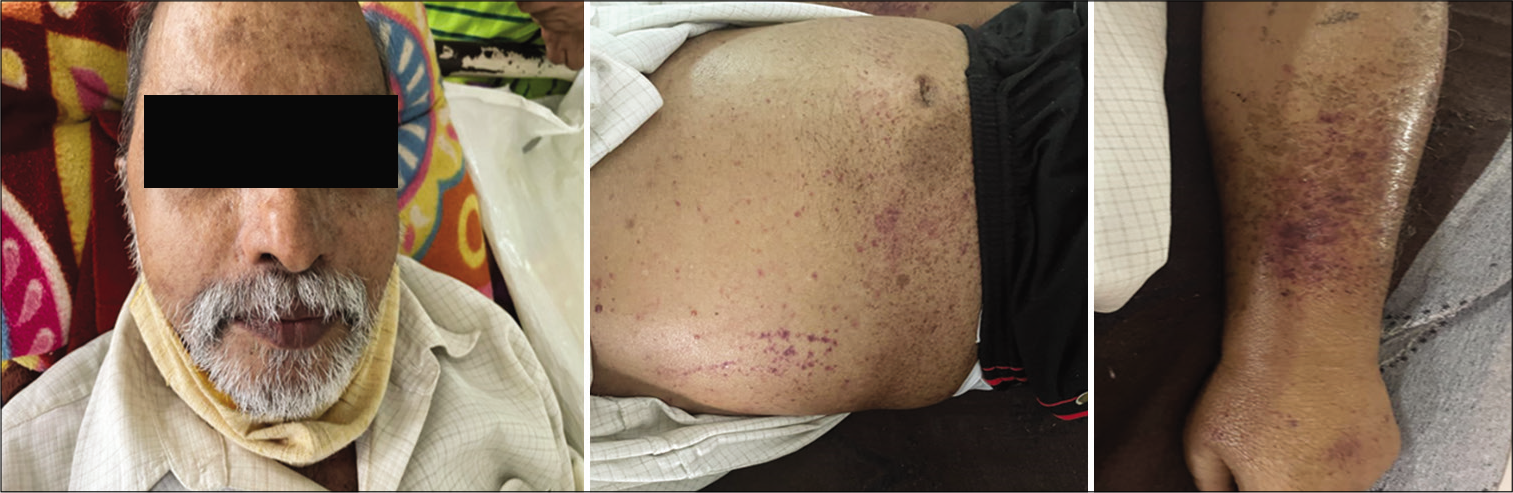
- Image showing periorbital pigmentation and purpuric spots.
The patient was started on injection human albumin and antibiotics. His repeat 2D echo was done which showed concentric left ventricular hypertrophy, severe tricuspid regurgitation, bilateral atrial enlargement and Grade 3 diastolic dysfunction (EF-60%), which was suggestive of restrictive cardiomyopathy. A strain 2D echo was done which showed cherry on top appearance, relative apical sparing and a GS score of –6 [Figures 2-5]. Repeat bone marrow aspiration revealed evidence of megaloblastic anaemia with 2% plasma cells. X-rays of skull and vertebrae were done which were normal. Urinary electrophoresis did not show any evidence of ‘M’ band. Serum protein profile by high-resolution agarose gel electrophoresis revealed the presence of a faint non-discrete protein band in gamma-globulin region; immunofixation electrophoresis identifies the very faint non-discrete protein band in gamma-globulin region of lambda lane corresponding to suspicious ‘M’ spike. A diagnosis of monoclonal gammopathy with AL amyloidosis with skin, cardiac and renal involvement was thus established.
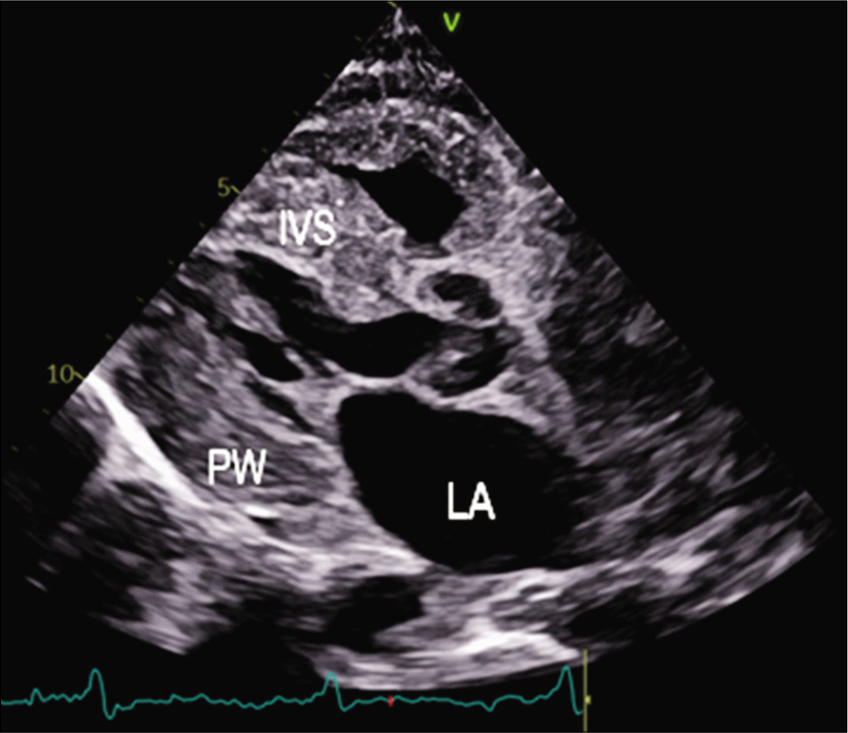
- PLAX view 2D echo showing thickened IVS.
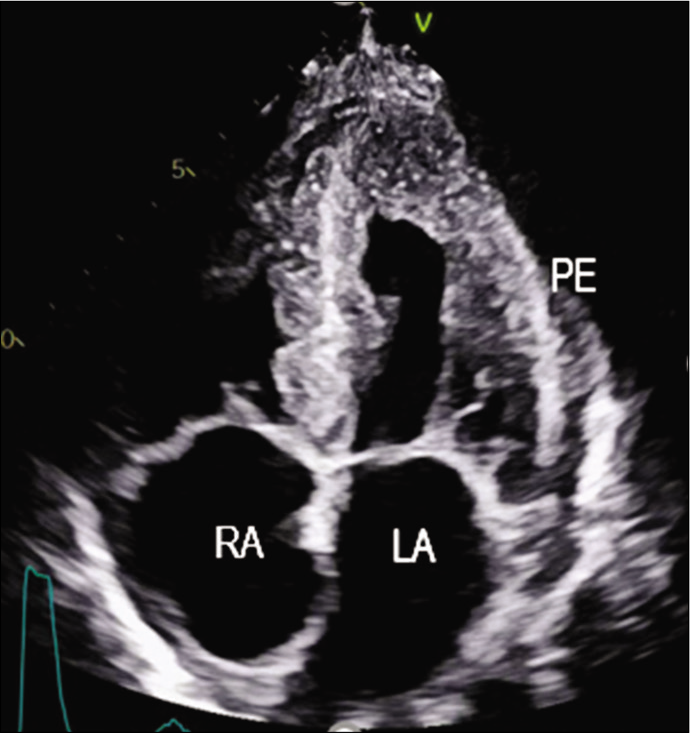
- Apical four-chamber view showing granular sparkling myocardium.
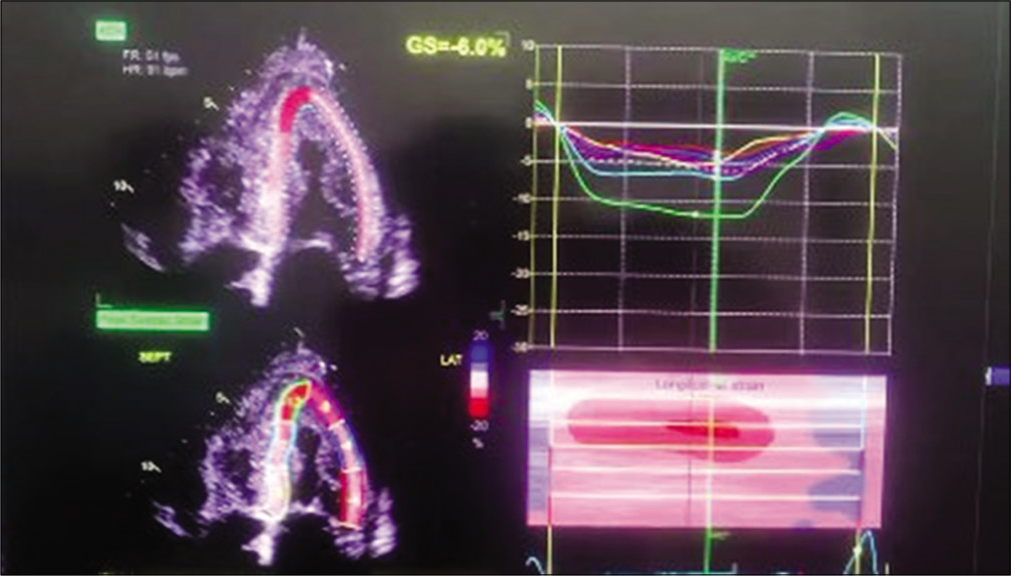
- Strain 2D echo showing speckle tracking and apical sparing.
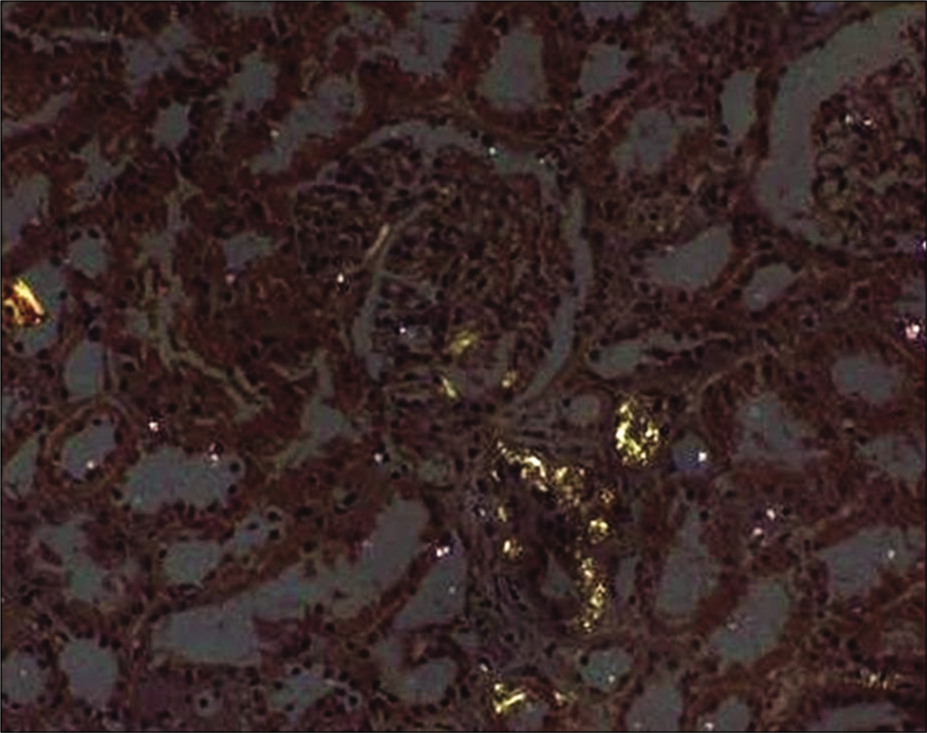
- Kidney biopsy showing Congo red staining.
Haematologist opinion was sought and the patient was started on injection bortezomib and cyclophosphamide along with other conservative management, unfortunately, we lost the patient during the course of the treatment.
DISCUSSION
Amyloidosis is a generic term that refers to the extracellular tissue deposition of fibrils composed of low-molecular-weight subunits of a variety of normal serum proteins. The median age at diagnosis is 64 years and <5% of patients are under the age of 40.[1-5] AL amyloidosis is a clonal plasma cell proliferative disorder in which fibrils of monoclonal light chains are deposited in the kidneys, heart and other tissues.[6] Affected patients may have AL amyloidosis alone or in association with other plasma cell dyscrasias.
The clinical presentation in AL amyloidosis depends on the number and nature of the organs affected. Non-specific systemic symptoms, including fatigue and unintentional weight loss, are common. Other common clinical presentations include nephrotic syndrome, restrictive cardiomyopathy, peripheral neuropathy and hepatomegaly with elevated liver enzymes.[7] Other less common but suggestive signs are macroglossia, purpura and an unexplained bleeding diathesis.
Diagnosis can be done based on international myeloma working group diagnostic criteria for systemic AL amyloidosis-[8]
Diagnosis of systemic AL amyloidosis requires all of the following
Presence of an amyloid-related systemic syndrome (e.g., renal, liver, heart, gastrointestinal tract or peripheral nerve involvement)
Positive amyloid staining by Congo red in any tissue (e.g., fat aspirate, bone marrow or organ biopsy)
Evidence that amyloid is light chain related established by direct examination of the amyloid using mass spectrometry-based proteomic analysis or immunoelectron microscopy
Evidence of a monoclonal plasma cell proliferative disorder (serum or urine monoclonal protein, abnormal-free light chain ratio or clonal plasma cells in the bone marrow).
AL amyloidosis should be distinguished from other forms of amyloidosis, from localised amyloidosis and from other types of monoclonal immunoglobulin deposition diseases (MIDD) because the clinical course and therapy are markedly different.[9] Other forms of amyloidosis will demonstrate staining with Congo red (a characteristic of amyloid), but direct examination of the amyloid material will not reveal immunoglobulin light chains (as seen in AL amyloidosis). Other forms of MIDD will have evidence of a monoclonal plasma cell proliferative disorder and many will demonstrate light chains in the serum. However, AL amyloidosis is the only MIDD that will demonstrate staining with Congo red.
AL amyloidosis: Treatment
The main treatment option in patients with light-chain (AL) amyloidosis is chemotherapy
A variety of regimens is used, including high-dose melphalan with autologous haematopoietic stem cell transplantation
Patients with poor performance status, major comorbidities, involvement of three or more organs and advanced cardiac amyloidosis are not considered transplant candidates
Bortezomib-based regimens (cyclophosphamide + Bortezomib + Dexamethasone) are first-line therapy for most patients who are not candidates for haematopoietic stem cell transplantation, even in patients with advanced cardiac disease (New York Heart Association [NYHA] functional Class III or IV)[10]
Loop diuretics are a mainstay of the treatment of cardiac amyloidosis
Beta-blockers, angiotensin-converting enzyme inhibitors and calcium channel blockers are contraindicated in amyloid cardiomyopathy
Anticoagulation is recommended in patients with amyloid cardiomyopathy with atrial fibrillation, intracardiac thrombi or an embolic event
The efficacy of implantable cardioverter-defibrillator therapy in patients with severe cardiac amyloidosis is unclear.
CONCLUSION
AL amyloidosis has a poor long-term prognosis when detected at an advanced stage
Earlier diagnosis is associated with lower early mortality and improved survival
Staging systems that incorporate NT-proBNP and cardiac troponin are used to prognosticate the disease.
Declaration of patient consent
Consent of Legally authorised Representative has been taken.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79:1817.
- [CrossRef] [PubMed] [Google Scholar]
- Amyloidosis (AL) Clinical and laboratory features in 229 cases. Mayo Clin Proc. 1983;58:665-83.
- [Google Scholar]
- Primary systemic amyloidosis: Multivariate analysis for prognostic factors in 168 cases. Blood. 1986;68:220.
- [CrossRef] [PubMed] [Google Scholar]
- Nationwide survey of 741 patients with systemic amyloid light-chain amyloidosis in Japan. Intern Med. 2018;57:181.
- [CrossRef] [PubMed] [Google Scholar]
- A modern primer on light chain amyloidosis in 592 patients with mass spectrometry-verified typing. Mayo Clin Proc. 2019;94:472-83.
- [CrossRef] [PubMed] [Google Scholar]
- Amyloid typing by mass spectrometry in clinical practice: A comprehensive review of 16, 175 samples. Mayo Clin Proc. 2020;95:1852-64.
- [CrossRef] [PubMed] [Google Scholar]
- Primary systemic amyloidosis: Clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45-59.
- [Google Scholar]
- International Myeloma working group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538.
- [CrossRef] [Google Scholar]
- Recognition of congestive heart failure due to senile cardiac amyloidosis. Biomed Pharmacother. 1989;43:101-6.
- [CrossRef] [Google Scholar]
- Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021;385:46-58.
- [CrossRef] [PubMed] [Google Scholar]




